


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
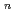 exciplexes
exciplexes高柳 敏幸; 志賀 基之
Physical Chemistry Chemical Physics, 6(13), p.3241 - 3247, 2004/07
被引用回数:29 パーセンタイル:67.75(Chemistry, Physical)300個のヘリウム原子からなるクラスターに付着したカリウム原子の光吸収ダイナミクスを最近われわれが開発した非断熱ハイブリッド量子シミュレーション法を用いて理論的に調べた。この方法では、カリウム原子のP電子を量子的に取り扱い、ヘリウム原子の運動は半古典経路積分セントロイド分子動力学法によって取り扱う。数十個のトラジェクトリーを計算した結果、初期励起の種類やクラスター初期構造に依存せず、すべての場合でカリウム原子がクラスターから脱離することがわかった。K*Heエキシマーの生成メカニズムを調べるため、ダイナミクスが断熱ポテンシャル面上で起こると仮定した計算も行い、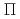 型の電子励起を行った場合にエキシマーが生成することを見いだした。これらの計算結果は過去の実験結果と定性的に一致する。また、エキシマー中のヘリウムの溶媒和構造を経路積分分子動力学法によって調べ、第一励起状態では6個のヘリウム原子が第一溶媒和相に束縛されるのに対し、第二励起状態では2個だけが束縛されることがわかった。
型の電子励起を行った場合にエキシマーが生成することを見いだした。これらの計算結果は過去の実験結果と定性的に一致する。また、エキシマー中のヘリウムの溶媒和構造を経路積分分子動力学法によって調べ、第一励起状態では6個のヘリウム原子が第一溶媒和相に束縛されるのに対し、第二励起状態では2個だけが束縛されることがわかった。
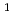 D)+N
D)+N O
O NO+NO
NO+NO河合 信之輔*; 藤村 陽*; 梶本 興亜*; 高柳 敏幸
Journal of Chemical Physics, 120(14), p.6430 - 6438, 2004/04
被引用回数:8 パーセンタイル:25.15(Chemistry, Physical)O(D)+NO反応で生成するNO(v=0,1,2)の回転状態の分布を測定した。回転温度はおよそ20000Kであり、分布は位相空間理論で予想されるものに近いことがわかった。この結果は、反応中間体の寿命がそれほど長くはないが、分布はほぼ統計的であることを意味する。しかしながら、回転量子数の大きな場合には、分布は位相空間理論で予想されるよりも早く減衰した。このことを理解するため、分子軌道計算に基づいたポテンシャル曲面を用いて古典軌道計算を行った。その結果、実験で得られた高い回転量子数の分布が反応出口領域のポテンシャルの影響を強く受けることがわかった。
D)+N(X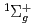 )O(
)O( P)+N(X) spin-forbidden electronic quenching collision
P)+N(X) spin-forbidden electronic quenching collision高柳 敏幸
Journal of Physical Chemistry A, 106(19), p.4914 - 4921, 2002/05
被引用回数:19 パーセンタイル:51.36(Chemistry, Physical)O(D)+N(X)O(P)+N(X)スピン禁制衝突について量子散乱計算を行った。一重項及び三重項についてそれぞれ一枚のポテンシャル面に簡略化した。標準的な堅密結合法により、電子的非断熱消光確率及び消光断面積を計算した。ポテンシャル面及びスピン軌道相互作用については過去にほかの研究者によって作製されたものを用いた。堅密結合の計算結果は、電子的非断熱過程が量子力学的な共鳴状態を経由して起こることを示した。このことは、これまで提唱されてきたポテンシャル交差モデルが、この非断熱過程には使えないことを示している。また、堅密結合の結果を半古典的ホッピングトラジェクトリー近似法による計算結果とも比較し、この近似が不十分であることも示した。
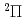 )+N(X)
)+N(X)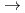 HCN(X
HCN(X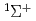 )+N(
)+N(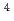 S) reaction
S) reaction和田 晃; 高柳 敏幸
Journal of Chemical Physics, 116(16), p.7065 - 7072, 2002/04
被引用回数:10 パーセンタイル:30.24(Chemistry, Physical)スピン禁制反応 CH(X)+N(X)HCN(X)+N(S) について、量子散乱理論を用いた計算を行った。CH分子を一個の原子とみなすことによって、自由度を3次元に落とした。分子軌道計算を用いて、スピン2重項及び4重項それぞれのポテンシャルエネルギー曲面を作製した。また、スピン軌道相互作用については過去の理論計算を用いた。超球座標を用いた堅密結合方程式を数値的に解いて、総反応確率を計算した。計算された確率は典型的な共鳴構造を示した。得られた確率から反応速度定数を計算し、実験結果と比較したところ、100倍ほど小さな値が得られたが、速度定数はスピン軌道相互作用に大きく依存することがわかった。
高柳 敏幸; 和田 晃
Chemical Physics Letters, 352(1-2), p.91 - 98, 2002/01
被引用回数:42 パーセンタイル:77.35(Chemistry, Physical)ヘリウムを含んだ化合物であるHHeF分子について、多配置参照配置間相互作用レベルの高精度分子軌道計算を行った。計算の結果、直線分子H-He-Fは準安定で、H-He-FH+He+Fの解離に対して0.224eVのエネルギー障壁をもち、H-He-FHe+HFの反応に対しては、0.448eVのエネルギー障壁をもつことがわかった。分子軌道計算を約3000点について行い、グローバルなポテンシャルエネルギー曲面を補間法によって作製した。そのポテンシャル面を使って3次元の時間に依存した波束計算を行ったところ、準安定共鳴状態の寿命は157fsと見積もられた。
 anion
anion高柳 敏幸; 和田 晃
Chemical Physics Letters, 348(1-2), p.514 - 520, 2001/11
被引用回数:14 パーセンタイル:41.25(Chemistry, Physical)時間に依存しない量子反応性散乱理論を用いてF(HD)アニオンの光電子脱離スペクトルの計算を行った。StarkとWernerの作製した高精度のポテンシャル面を使った。計算したFHD及びFDH両アニオンのスペクトルには、束縛回転に相当するブロードなピークがいくつか見られた。これは、以前研究されたFH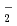 のスペクトルで見られたものと本質的に同じである。さらに、FHDアニオンでは、遷移状態共鳴に相当するピークが見られた。これは、最近、詳細な反応断面積の測定によって実験的に見出されているものである。本理論計算結果は光電子脱離スペクトル実験によって、遷移状態共鳴が見出される可能性があることを強く示唆するものである。
のスペクトルで見られたものと本質的に同じである。さらに、FHDアニオンでは、遷移状態共鳴に相当するピークが見られた。これは、最近、詳細な反応断面積の測定によって実験的に見出されているものである。本理論計算結果は光電子脱離スペクトル実験によって、遷移状態共鳴が見出される可能性があることを強く示唆するものである。
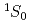 )+HF(
)+HF(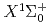 )He()+H(
)He()+H(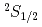 )+F(
)+F(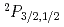 )
)高柳 敏幸; 和田 晃
Journal of Chemical Physics, 115(14), p.6385 - 6393, 2001/10
被引用回数:5 パーセンタイル:15.25(Chemistry, Physical)衝突誘起解離反応He()+HF()He()+H()+F()について3次元半古典理論を用いて、スピン軌道相互作用による励起状態の寄与について検討した。衝突動径座標を古典力学で取り扱い、HFの振動と回転は量子力学的に取り扱った。ポテンシャルエネルギー曲面は、Diatomics-in-Molecule (DIM)近似を使って計算した。その結果、生成するF原子のうち、約20%程度がスピン軌道励起状態に分布することを見いだした。この結果は解離過程が基底状態で起こるとする従来の理論に修正を迫るものである。さらに、電子的非断熱遷移はH原子とF原子が十分離れた位置で起こるため、異方性の効果は小さいことがわかった。
D)+NONO+NO reaction高柳 敏幸; 和田 晃
Chemical Physics, 269(1-3), p.37 - 47, 2001/07
被引用回数:14 パーセンタイル:41.25(Chemistry, Physical)O(D)+NONO+NO反応について、量子反応性散乱計算を行った。ポテンシャルエネルギー曲面は、CASPT2レベルの高精度の分子軌道計算を行い、解析関数にフィットして作製した。反応側及び生成側の配向角を固定したモデルを用いることによって、次元を3次元に落とした。この反応では2種類のNO分子が生成する。反応熱はおもに新しく生成するNO分子の振動に分配されるが、もともと存在したNO振動モードにも、ある程度エネルギーが分配されることを見出した。このことはもともと存在したNO結合が、必ずしもスペクテータではないことを示している。
 HF and HDF van der Waals molecules
HF and HDF van der Waals molecules高柳 敏幸; 和田 晃
Chemical Physics Letters, 338(2-3), p.195 - 200, 2001/04
被引用回数:15 パーセンタイル:43.4(Chemistry, Physical)ファン・デル・ワールス分子の前期反応過程,D…HF+h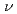 →DH+F及びH…DF+h→HD+Fについて理論的に検討した。超球座標を用いた3次元の時間に依存しない量子反応性散乱理論を使って計算を行った。また正確な分子軌道計算によって作成されたポテンシャルエネルギー曲面を用いている。その結果、水素原子移動反応であるD…HF+h→DH+F過程は非常に大きな確率で起こるが重水素原子の移行するH…DF+h→HD+F反応の確率は極めて小さいことがわかった。このことは前期反応過程ではトンネル効果が支配的な役割をすることを示している。
→DH+F及びH…DF+h→HD+Fについて理論的に検討した。超球座標を用いた3次元の時間に依存しない量子反応性散乱理論を使って計算を行った。また正確な分子軌道計算によって作成されたポテンシャルエネルギー曲面を用いている。その結果、水素原子移動反応であるD…HF+h→DH+F過程は非常に大きな確率で起こるが重水素原子の移行するH…DF+h→HD+F反応の確率は極めて小さいことがわかった。このことは前期反応過程ではトンネル効果が支配的な役割をすることを示している。
 P
P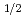 )+H HBr+H reaction
)+H HBr+H reaction高柳 敏幸; 黒崎 譲
Journal of Chemical Physics, 113(17), p.7158 - 7164, 2000/11
被引用回数:43 パーセンタイル:77.61(Chemistry, Physical)スピン軌道相互作用による電子的非断熱遷移を伴う反応、Br(P)+H HBr+Hについて3次元量子反応性散乱計算を2つの計算方法を用いて行った。1つは超球座標を用いたclose-coupling法で、もう一方は、虚数の吸収ポテンシャルを用いた一般化R行列伝播法である。後者では反応側のJacobi座標を用いた。ポテンシャル曲面としてはTruhlarらによる(2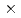 2)のdiabaticなポテンシャル曲面を用いた。いずれの方法でも数値的に十分収束した計算結果を得ることができた。また、得られた結果から電子的非断熱遷移が反応の入口でほとんど起こるが、その効率は小さいことがわかった。
2)のdiabaticなポテンシャル曲面を用いた。いずれの方法でも数値的に十分収束した計算結果を得ることができた。また、得られた結果から電子的非断熱遷移が反応の入口でほとんど起こるが、その効率は小さいことがわかった。
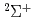 ) with CH, CH
) with CH, CH , HO and their isotopic variants
, HO and their isotopic variants梅本 宏信*; 寺田 直樹*; 田中 邦和*; 高柳 敏幸; 黒崎 譲; 横山 啓一
Chemical Physics, 259(1), p.39 - 47, 2000/09
被引用回数:9 パーセンタイル:27.42(Chemistry, Physical)NO分子の第一励起状態Aとアセチレン,エチレン及び水との反応において、水素原子が直接生成することを初めて実験的に確認した。水素原子のドップラー分光の結果からアセチレンと水については1/4のエネルギーが並進運動に分配され、エチレンについては1/7であった。この結果は反応過程で極めて寿命の短い中間体が生成していることを示すものである。反応のメカニズムをさらに詳細に理解するため、ab initio分子軌道法によるポテンシャルエネルギー曲面の計算を行った。
D)+CH reaction; Does the N(D) atom really insert into CH bonds of alkane molecules?高柳 敏幸; 黒崎 譲; 横山 啓一
International Journal of Quantum Chemistry, 79(3), p.190 - 197, 2000/09
被引用回数:11 パーセンタイル:49.22(Chemistry, Physical)最近、米国の量子化学研究者によってN(D)原子がメタンのCH結合に挿入しないことが報告されたが、本論文はその研究結果に対する反論である。多配置ハートリーフォック計算、さらに大規模な配置間相互作用を考慮した計算によって、N(D)原子がCH結合に挿入してCH NH(A'')を生成することを改めて理論的に示した。さらに興味深いことに二重項第一励起状態のポテンシャル曲面上でも挿入反応が起こることを見いだした。この場合はCHNH(A')分子が生成する。これらの結果は最近われわれが行ったN(D)+Hのポテンシャル曲面の結果とよく似ている。
NH(A'')を生成することを改めて理論的に示した。さらに興味深いことに二重項第一励起状態のポテンシャル曲面上でも挿入反応が起こることを見いだした。この場合はCHNH(A')分子が生成する。これらの結果は最近われわれが行ったN(D)+Hのポテンシャル曲面の結果とよく似ている。
Balucani, N.*; Algia, M.*; Cartechini, L.*; Casavecchia, P.*; Volpi, G. G.*; 佐藤 圭*; 高柳 敏幸; 黒崎 譲*
Journal of the American Chemical Society, 122(18), p.4443 - 4450, 2000/05
被引用回数:77 パーセンタイル:87.92(Chemistry, Multidisciplinary)第一励起状態であるN(D)原子のアセチレンとの反応について、公差分子線と高いレベルの分子軌道計算によって調べた。主たる反応メカニズムはN(D)+CHHCCN+Hであり、窒素と水素原子が交換する。この反応はタイタンの大気化学に非常に重要であることが予想される。これまで大気中のCHを含んだ化合物はほとんどイオン分子反応で生成すると考えられていたが、本研究は中性分子間の反応も重要であることを示す。
S, D, P)+H reactions高柳 敏幸; 黒崎 譲; 横山 啓一
Chemical Physics Letters, 321(1-2), p.106 - 112, 2000/04
被引用回数:24 パーセンタイル:58.71(Chemistry, Physical)多配置参照配置間相互作用の方法を用いた分子軌道法によってN(S, D, P)+Hの反応のポテンシャルエネルギー曲面を計算した。特にC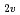 とCoor配置の計算を重点的に行い、2次元のポテンルシャル面を詳細に作製した。その結果、N(D)+H反応については5枚のポテンシャル面のうち、2枚が重要であることを明らかにした。またこれらのポテンシャル面が最低4重項のポテンシャルと交差し、N(S)+Hへの非断熱遷移が起こりうることを見いだした。また計算結果に基づき、N(P)+Hの消光過程のメカニズムについて検討した。
とCoor配置の計算を重点的に行い、2次元のポテンルシャル面を詳細に作製した。その結果、N(D)+H反応については5枚のポテンシャル面のうち、2枚が重要であることを明らかにした。またこれらのポテンシャル面が最低4重項のポテンシャルと交差し、N(S)+Hへの非断熱遷移が起こりうることを見いだした。また計算結果に基づき、N(P)+Hの消光過程のメカニズムについて検討した。
) reaction system
reaction system高柳 敏幸; 黒崎 譲; 市原 晃
Journal of Chemical Physics, 112(6), p.2615 - 2622, 2000/02
被引用回数:62 パーセンタイル:86.41(Chemistry, Physical)非断熱遷移を伴う(D+H)イオン分子反応について3次元量子散乱計算を行った。超球座標を使った時間に依存しないclose-coupling法を用いた。ポテンシャルエネルギー曲面として(33)のDIMポテンシャルを使った。正確な量子論の計算結果を半古典的なトラジェクトリホッピングの結果と比較した。その結果Tullyによって提唱されている方法のほうが従来から使われているTully-Prestonの方法よりも量子論の結果をよく再現することがわかった。これはTully-Prestonの方法が、ポテンシャルの交差付近でのみの電子遷移しか考慮していないことが原因である。
…HD van der Waals molecule高柳 敏幸; 黒崎 譲
Physical Chemistry Chemical Physics, 2(4), p.665 - 670, 2000/02
被引用回数:10 パーセンタイル:30.33(Chemistry, Physical)赤外励起によって引き起こされるファン・デル・ワールス分子の前期反応過程、H…HD+hH+Dについて反応性散乱理論を使った理論的研究を行った。正確な分子軌道計算をもとにして作製されたStarck-Meyerのポテンシャルエネルギー曲面を用いた。その結果H…HD(=1)という共鳴状態を経ると、反応が5 10%の確率で起こることが予想された。また回転励起に関する共鳴状態を経由した場合、ほとんど前期解離過程H…HD+hH+HDが起こることがわかった。この結果はH
10%の確率で起こることが予想された。また回転励起に関する共鳴状態を経由した場合、ほとんど前期解離過程H…HD+hH+HDが起こることがわかった。この結果はH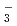 ポテンシャル曲面においては回転非断熱遷移の確率が大きいことを意味している。
ポテンシャル曲面においては回転非断熱遷移の確率が大きいことを意味している。
S and DS佐藤 圭*; 若林 成二*; 松原 孝*; 杉浦 円*; 綱島 滋*; 黒崎 譲*; 高柳 敏幸
Chemical Physics, 242(1), p.1 - 10, 1999/00
被引用回数:7 パーセンタイル:22.54(Chemistry, Physical)CH+HS,CD+HS,CH+DS及びCD+DS反応の295Kでの反応速度定数をレーザー誘起けい光法を用いて測定した。同位体効果はたいへん小さいことがわかった。反応のメカニズムを高いレベルの分子軌道計算結果から検討した。その結果CHラジカルはHS中のS原子にバリヤーなしで付加するのが初期過程であることがわかり、実験結果を強く裏付けるものであった。またRRKM計算により、生成物の分岐比についても検討した。
D)+acetylene reaction高柳 敏幸; 黒崎 譲*; 横山 啓一; 佐藤 圭*; 綱島 滋*
Chemical Physics Letters, 312(5-6), p.503 - 510, 1999/00
被引用回数:9 パーセンタイル:28.09(Chemistry, Physical)分子軌道計算結果を用いて、N(D)+CH,CD反応の反応速度定数の変分的遷移状態理論計算を行った。計算値と実験値の比較から、量子力学的効果である非断熱過程が重要であることを明らかにした。さらにこのことを半定量的に確かめるために、正確な分子軌道理論を用いて、長距離領域でのポテンシャルエネルギー曲面の計算を行った。その結果ファンデルワールス領域でポテンシャルの交差が起こっていることがわかった。
D)+CH insertion reaction高柳 敏幸; 黒崎 譲*
Journal of Molecular Structure; THEOCHEM, 492, p.151 - 158, 1999/00
N(D)+CH反応について分子軌道法を直接用いた古典的トラジェクトリー計算を行った。反応の遷移状態から出発し、反応座標方向にエネルギーを与えて計算を行った。N(D)原子がCHのCH結合に挿入して生成するCHNH中間体ラジカルの寿命は非常に短いことがわかった。この結果は実験結果と定性的に一致する。このことからエネルギーはおもにCHとNHフラグメントの相対運動に分配されると予測することが可能である。
D) with CHOH and its isotopomers梅本 宏信*; 金剛 晃一*; 稲葉 重信*; 園田 保之*; 高柳 敏幸; 黒崎 譲
Journal of Physical Chemistry A, 103(35), p.7026 - 7031, 1999/00
被引用回数:8 パーセンタイル:25.43(Chemistry, Physical)メタノールとN(D)原子の反応経路をレーザー誘起けい光法とab initio分子軌道法を用いて調べた。NH及びOHラジカルが反応生成物として検出され、これらの分子の内部状態が統計的でないことが確認された。このことは反応中間体の寿命がエネルギーがランダムになる時間より短いことを示している。CHOD及びCDOHとの反応からNH,ND,OH及びODが生成物として検出された。測定された生成物の分岐比から、N原子はCH結合に挿入しその後水素原子のスクランブリングが起こると結論された。この結果は分子軌道法の計算結果と定性的に一致することがわかった。
